My laboratory is interested in the evolutionary history of Mycobacterium pathogens, specifically those that cause tuberculosis (TB) and leprosy. These diseases have plagued humans for millennia and continue to be a public health problem today. We are currently involved in three research projects that investigate (1) the ancient history of TB in the Americas, (2) the modern spread of leprosy in the Pacific, and (3) the prevalence of these diseases in animals and potential for zoonotic spillover.
The evolutionary history of tuberculosis using ancient DNA:
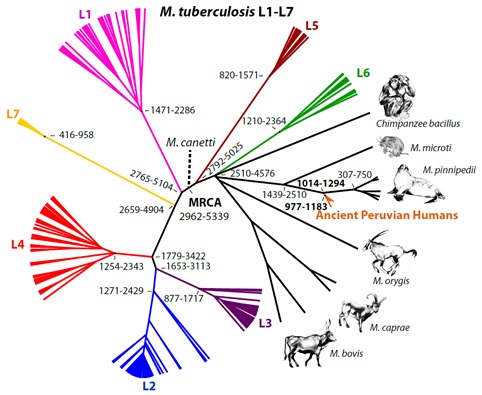
Tuberculosis (TB) has a long history of affecting humans with current estimates of its origins ranging from 3-6 million years ago. TB is caused by bacteria (specifically Mycobacterium tuberculosis and several closely related mycobacteria) that are found in people as well as in other animals. In our research, we found that strains of TB in ancient Peruvians living about 1000 year ago originally came from seals. The seal TB likely “jumped” from seals to humans during the process of butchering or from eating under-cooked meat. Interestingly, tests for disease in seals and sea lions find that only southern hemisphere seals and sea lions are affected by TB. Previous research of animal TB strains suggests that South African fur seals were infected with TB by another animal (perhaps a rodent). The seal TB then likely spread among seal species (interacting around Antarctica) until it infected species that range up along the coast of South America. Also, the genetic data from the ancient Peruvian TB suggests that these strains might have adapted so they could be transmitted better from human to human rather than just from seal to human. We are currently trying to understand how widespread the seal TB became in people in the Americas before European contact. We have new preliminary evidence that it spread north to the highlands of Columbia and on into Mexico, likely via human to human transmission. Today, human TB strains throughout the Americas are closely related to European strains indicating that the pre-contact seal-derived strains were replaced. We do not yet understand how quickly this occurred in different parts of the Americas (and whether there were multiple introductions of European TB strains after contact). A second aim of our research is to understand the sources and timing of the introduction of new strains after European contact. This research is in collaboration with Jane Buikstra and Kelly Blevins (Center for Bioarchaeological Research, SHESC, ASU), Tanvi Honap (University of Oklahoma), Kirsti Bos, Alex Herbig and Johannes Krause (Max Planck Institute for Evolutionary Anthropology). This work was funded by NSF BCS-0612222 (PI Stone, 2006-2010), BCS-1063939 (PI Stone, 2011-2015) and BCS-1945812 (PI Stone and Co-PI Blevins, 2020-2021) and by the Wenner Gren Foundation for Anthropological Research.
A Bioarchaeological Investigation of Mobility and Infectious Disease: The proposed project explores how tuberculosis spread within and among communities occupying the Osmore Drainage of southern Peru from 385 BCE to 1476 CE. We aim to address an enigma whereby basal ancient American pinniped Mycobacterium tuberculosis complex (MTBC) strains appear in the Valley of Mexico and elevated locations of Colombia, whereas chronologically earlier sites of the coastal Andes present evidence of derived strains. Using state-of-the-art molecular methods, we will test implications of a model whereby pinniped MTBC strains jumped to humans many times in South America, subsequently becoming a human disease that was carried north in South America and may have followed traders via sea routes to western Mexico and hence to the Valley of Mexico. Meanwhile, waves of transmission directly from sea mammals to human continued on the Andean coast. Focusing upon the rich archaeological resources of the Osmore Valley in the south-central Andes in present-day Perú, this model predicts that people from Osmore Valley Huaracane and Middle Horizon sites (Chen Chen, Omo, and Rio Muerto) should exhibit MTBC strains basal to those from Late Intermediate Chiribaya peoples in the lower and middle Osmore Valley. These strains will be more similar to the basal strains from later pre-contact Colombia and Tlatelolco, México than the derived, more recently introduced Chiribaya strains. Generating an infectious disease landscape that includes evidence of human mobility, interaction, ethnicity and dietary patterns will require investigations of light and radiogenic isotopes, building on the rich baseline data already available. Basing expectations on currently available evidence about MTBC outbreaks, we also focus upon strain diversity and evidence of individual mobility and community interactions. In developing a comprehensive understanding of the people who died with MTBC, this study will include a life history approach that integrates information about health with other evidence of disease, diet and funerary treatment. This life history information will serve as a basis for both the project’s research synthesis and its outreach program. In summary, this study will 1) thoroughly census skeletal MTBC presence in the Osmore River valley, Perú, a rich archaeological region where both skeletal and molecular evidence of MTBC have been documented; 2) extend knowledge of molecular MTBC signals by testing previously undocumented and samples and returning with Next Generation methods to samples we first studied using PCR techniques; 3) explore the presence of MTBC and other pathogens through shotgun sequencing of samples derived from dental pulp cavities; 4) investigate disease transfer patterning through the study of human mobility; and 5) consider the degree to which infected individuals were stigmatized, as interpreted through within-site funerary treatments
This project is in collaboration with Jane Buikstra, Kelly Knudson, Allisen Dahlstedt (ASU) and Kelly Blevins (University of Durham) and funded by NSF, BCS-2217953 (2022-2025).
OMID project introduction:
Modern leprosy and ancient migration in the Pacific:
Leprosy, or Hansen’s disease, is an infectious disease that has plagued humans for millennia. Discussions of the origin of Mycobacterium leprae, the pathogen that causes most human leprosy cases, have focused largely on Western Eurasia. This project seeks to expand that geographic and characterize the spread of this pathogen in the Pacific. Specifically, we will analyze DNA from clinical formalin-fixed paraffin-embedded (FFPE) tissue samples collected from the Pacific Islands in the past three decades. Preliminary results (see: Blevins et al. 2020) support a premodern introduction of M. leprae into the Pacific Islands, which would have been spread during multiple waves of migration throughout the Pacific. Current work is seeking to fine tune our understanding of this geographic spread with spatial analyses of between island transmission. We are also seeking to improve methods to extract and sequence DNA from FFPE samples, as the tissue preservation process often results in fragmented, degraded DNA. This work is in collaboration with Dr. Keolu Fox (University of California, San Diego), Dr. Christopher Lum (John A Burns School of Medicine, University of Hawaii), and Dr. Kanako Furuta (Hawaii Pathologists Laboratory).
Disease spillover in the Amazon
Disease exchange between humans and other animals has been an important adaptive pressure during our evolutionary history, as well as a source of emerging infection diseases in our modern age. It is likely that non-human primates and other mammals have long been important sources of new pathogens because of their close evolutionary relationship to us. The biological similarities mean that pathogens require few changes to make the “jump” to humans. We are investigating the infectious disease exchange between human and local mammals using the causative agents of leprosy – Mycobacterium tuberculosis and M. leprae – as case studies. Zoonotic spillover of leprosy from armadillos to humans has been well documented in the Americas, with drug resistant isolates found in South America. Our project has two major components: (1) test samples from a wide range of mammals in endemic regions for the presence of these pathogens and (2) use data generated to understand evolutionary changes required to enable spillover. An understanding of the past and present interactions between mammals and mycobacteria will provide baseline data to inform current and future public health, conservation, and diagnostic/treatment efforts. This research is in collaboration with Dr. Luciano Nakazato (Universidade Federal de Mato Grosso) and Dr. Gideon Erkenswick (Field Projects International).